En esta aparición en el programa de divulgación científica Trending Ciencia, podéis escucharme comentando la noticia de hace unos días referente a la utilización de las tecnologías de clonación por parte del gobierno Chino dirigidas al ganado porcino en pos de la investigación genética. Además, os pondréis al tanto del potencial de este gigante asiático en el campo de la secuenciación y su objetivo de clonar lo que llaman «animales adorables».
No dudéis en comentar este episodio en esta misma entrada en la web o mediante las redes sociales mencionando nuestra cuenta oficial de Twitter @TrendingCiencia o personalmente al autor del programa, que soy yo,@DoctorGenoma. O es de más animar a todo el que le guste la ciencia a suscribirse a nuestro podcast mediante su FEED o a través de las plataformas iTunes e iVoox.
Los sistemas de secuenciación actuales están permitiendo el avance en el conocimiento de todos los organismos de interés científico. En esta ocasión, el blanco era una planta muy especial por su importancia evolutiva: el loto sagrado.
El loto sagrado (Nelumbo nucifera) es un símbolo de la pureza espiritual y la longevidad. Sus semillas pueden sobrevivir hasta 1.300 años, sus pétalos y hojas repelen la suciedad y el agua, y sus flores generan calor para atraer a los polinizadores.
Ahora, investigadores de la Universidad de Illinois han publicado en la revista Genome Biology el trabajo de secuenciación de su genoma y los resultados ofrecen información sobre algunos de sus misterios. La secuencia revela que de todas las plantas secuenciados hasta el momento el loto sagrado tiene el parecido más cercano al ancestro de todas las eudicotiledóneas, una categoría amplia de plantas con flores en la que se incluyen la manzana, la col, el cactus, la planta del café, el algodón, la uva, el melón, el álamo, la soja, la girasol, el tabaco y el tomate por ejemplo.
«El loto sagrado carece de una zona del genoma que se encuentra triplicada en la mayoría de los otros miembros de esta familia», dijo la doctora Ray Ming del Instituto de Biología Genómica de la Universidad de Illinois, que dirigió el análisis con Jane Shen-Miller, profesor de biología de plantas de la Universidad de California en Los Angeles (que germinó una semilla de loto sagrado de 1.300 años de antigüedad), y Shaohua Li, director del Jardín Botánico de Wuhan en la Academia China de Ciencias.
La aparición de duplicaciones o más copias de ciertas zonas del genoma es muy importante desde el punto de vista evolutivo. Algunos de los genes duplicados (o con más copias) conservan su estructura y función original, por lo que producen más de un producto del gen, como una proteína, por ejemplo. Si estos cambios son beneficiosos, los genes persisten, si son perjudiciales, desaparecen del genoma.
«Muchos cultivos agrícolas se benefician de las duplicaciones del genoma, incluyendo el banano, la papaya, la caña de azúcar, la fresa, la sandía y el trigo», dijo Robert Van Buren, un estudiante graduado en el laboratorio y colaborador de Ming en el estudio.
El loto sagrado experimentó una duplicación independiente a sus «parientes cercanos» en la rama inicial de las eudicotiledóneas que ocurrió hace unos 65 millones de años, según los investigadores. Y se ha mantenido una gran proporción de esos genes duplicados (alrededor del 40 por ciento).
Los investigadores han encontrado evidencia de que se retuvieron la duplicación de genes relacionados con la formación de cera (que permite a la planta repeler el agua y mantenerse limpia) y que permita su supervivencia en ciertos hábitats, por ejemplo.
Al observar los cambios en la duplicación de genes, los investigadores encontraron que el loto tiene una tasa de mutación lenta en relación con otras plantas. Estas características hacen de la loto una planta de referencia ideal para aumentar el conocimiento de otras eudicototiledóneas.
Tras algún intento de probar la supercomputación para mi tesis doctoral, finalmente he podido tener acceso al servicio prestado por la Fundación del Centro de Supercomputación de Castilla y León donde pude disfrutar de darle a los comandos para usar algún que otro core de Caléndula, que así se llama el «monstruo» en cuestión.
Parte interior de uno de los clústeres de cálculo de Caléndula. Pulsar para ampliar.
El curso trataba de los análisis de metagenomas mediante supercomputación con mucha temática dirigida al uso de la ultrasecuenciación. Como ya les comenté a los organizadores, es una pena que exista una muy deficiente base informática en los investigadores de biología molecular. De los 19 presentes, yo era el único que sabía teclear en condiciones instrucciones en UNIX. Pero seguro que, al menos los asistentes, espabilarán. Casi todos mostraron interés por probar de una vez alguna distribución de Linux.
Pasando al meollo, la verdad es que para un friki de la tecnología como yo es un curso muy recomendable. A parte del título que te brinda por un precio nada despreciable de 350€ (más que ajustado ante los demás cursos ofertados sobre temas bioinformáticos a nivel nacional), la experiencia de meterse de lleno en análisis de secuencias genómicas y metagenómicas procedentes de distintos sistemas de próxima generación como los Roche 454 o los HiSeq de Illumina es apasionante. Trabajar de forma práctica siguiendo todos los pasos como si fuera un proyecto real abre mucho la mente ante todo el trabajo que hay detrás de un análisis de este estilo. Eso y que no estoy sólo en esto del frikismo bioinformático.
Hubo una primera parte en la que Jesús Lorenzana, uno de los responsables directos de Caléndula, nos dio unas nociones técnicas del proceso del sistema de supercomputación instalado en el CRAI-TIC de la Universidad de León. Datos técnicos que para algunos sólo servirán para aburrir pero que quitan el hipo cuando te has pasado años montando equipos.
Esquema de la disposición y caracterísiticas del Supercomputador Caléndula. Pulsar para ampliar.
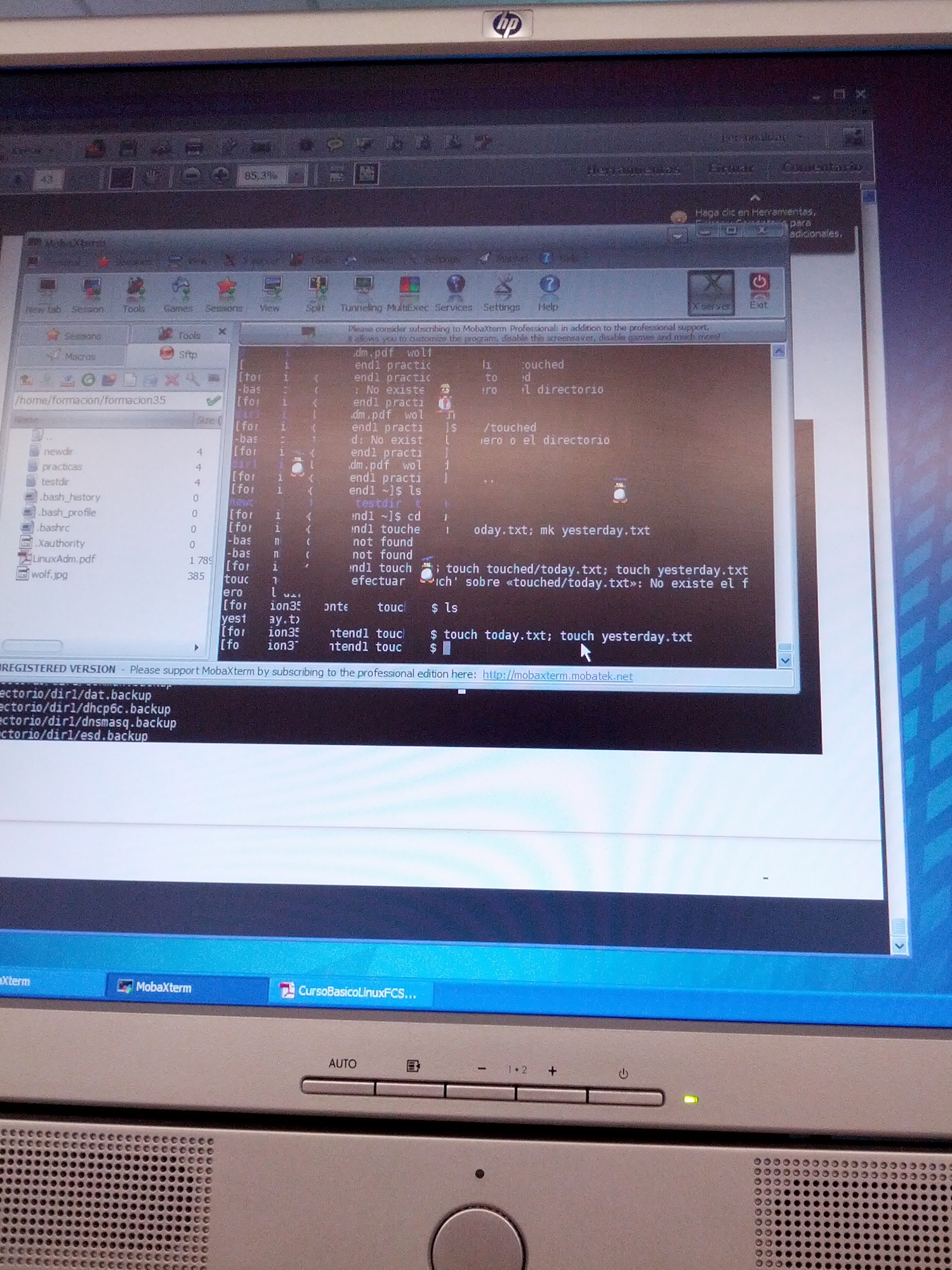
Después Mariví López nos dio las nociones básicas para manejarse en condiciones en el entorno Linux. Cosas simples, sí, pero que incluso a un autodidacta como yo vienen bien recordar de vez en cuando. Muchas de las órdenes que he dado a través del terminal las he tecleado por inercia sin saber específicamente alguno de los atributos. Pero bueno, también son más de 12 años aprendiendo a fuerza bruta y me vino genial. Algunos comandos que luego se necesitaron no se pudieron dar, pero no hubo problemas puesto que dichas órdenes fueron necesarias posteriormente en otra clase en la que la falta de tiempo no era lo primordial y se aprendieron sobre la marcha sin problemas.
Simpáticas animaciones del cliente para el acceso SSH MobaXterm. Pulsar para ampliar.
La tarde de ese primer día fue más relajada tras el «estrés» mañanero Jesús A. Gómez-Ochoa hizo una muy sensata introducción a la bioinformática preguntando al principio cuál era nuestra experiencia y que facilitó la orientación de la charla.
En la mañana del segundo día, Alexander Sánchez Pla introdujo los sistemas de secuenciación de próxima generación y nos mostró el uso de FastQC en modo gráfico, muy importante para comprender y comparar lo que tenemos delante tras pasar nuestro ADN por un Illumina.
Aunque todas las clases fueron amenas, en el momento en el que llegó la hora de picar comandos para analizar datos de verdad el caos se hizo presente. Posiblemente el profesor encargado de la partes de RNAseq creyó que todos tenían un background en el uso de linux lo suficientemente correcto como para no estar pensando más en la orden que dar que en el análisis en sí, en los pasos a dar. La ejecución de los scripts que es básica llegó como un jarro de agua fría para mis compañeros. Y es normal. Posiblemente en otras ediciones se baraje dar una formación algo más gradual dejando esta parte de RNAseq casi para el final. Todo también unido a que fue la última clase de la mañana y la falta de azúcar hizo estragos.
Las sesiones vespertinas posteriores fueron teóricas iniciándose con el análisis mediante el uso de Microarrays por Enrique J. de Andrés Galiana. Una ponencia que, desde mi punto de vista, fue muy matemática y en la que me sentí bastante perdido.
Características de cuatro supercomputadores punteros a nivel mundial. Pulsar para ampliar.
Javier Tamames siguió con su parte dedicada al análisis de la diversidad microbiana y de metagenómica explicando todos y cada uno de los pasos a seguir para cada tipo de experimento. Con una página web creada para el curso como apoyo tanto para él como para los alumnos y agilizando el aprendizaje correctamente, se intercalaron los usos de la supercomputación con herramientas vía web para análisis menos exigentes. La repetición de los pasos y la consecución de los análisis siguiendo esas instrucciones provocó una buena asimilación de estos estudios que se basaban en análisis de secuencias del 16S con sistemas Roche 454.
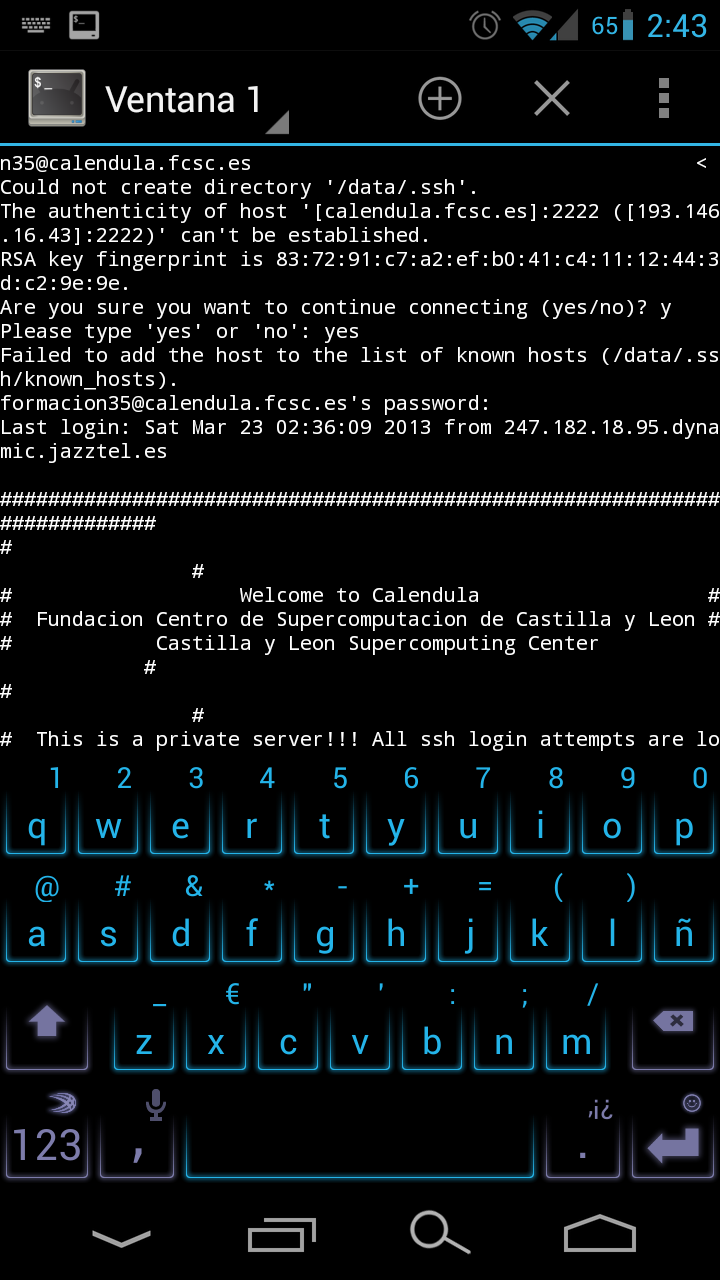
Las prácticas con el Dr. Tamames duraron hasta el último minuto del último día de clase mañanera, tras el cuál nos dispusimos a visitar el sistema de supercomputación Caléndula. Jesús nos desgranó, a nivel comprensible por todos, tanto los sistemas de emergencia como las tecnologías que componen el gigantesco puzzle de supercomputación que hasta hace poco se encontraba entre el Top 500 mundial.
Para que notéis la sensación de estar en el superordenador, os pongo a continuación el sonido ensordecedor grabado in situ.–>PULSAR AQUÍ para escuchar el sonido del supercomputador.
Accediendo a Caléndula desde Android. Pulsar para ampliar.
Aunque tras terminar el curso no se había podido asimilar todo el temario, la disponibilidad para descargarse todo lo impartido y tener acceso a Caléndula durante la semana siguiente (para mí poco tiempo debido a la cercanía de la Semana Santa y que soy un biólogo algo inquieto), provoca que se pueda exprimir al máximo el evento. Aunque para mí, una semana me supo a poco. Ojalá, en un futuro no demasiado lejano, todos los inquietos por estos sistemas de análisis se puedan dividir en categorías para saciar las distintas mentes. Pero al día de hoy es una buena experiencia que no está al alcance de muchos. Eso sí, después de ver las tripas de Caléndula se me va a hacer pequeño cualquier otro sistema informático convencional.
Imagen de uno de los días del curso.
Agradecimientos: quiero dar las gracias tanto a todos y cada uno de los instructores por su paciencia y buen hacer y en especial a Jesús Lorenzana y Ruth Alonso por la ayuda prestada para la recopilación de datos e imágenes para este artículo.
En el momento en el que me llegó la noticia vía Twitter, mi primera impresión fue de extrañeza ante el titular, ya que una de las cosas que se aprende en la carrera (y en mi caso la idea quedó más arraigada ya que me dedico a ello) es que el genoma es el conjunto de información genética que contiene un organismo en particular. Pero me importaba más leer el artículo científico y no di más importancia al tema al asociarlo a los medios de comunicación, que suelen buscar llamar la atención. En general, el titular era «se ha secuenciado el genoma de la leucemia».
Por falta de tiempo no pude más que leer el artículo del que se hablaba en todos los medios y me sumergí en él sin volver a pensar en ello más de lo que comenté con los de mi entorno el primer día. Sin embargo pude leer el artículo del blog La muerte de un ácaro (que recomiendo la suscripción) que relata perfectamente lo sucedido. Voy a citar la explicación sencilla de por qué no es correcta la afirmación de «secuenciar el genoma de la leucemia» (extracto del artículo del blog «La muerte de un ácaro»)
[…]Secuenciar es obtener el orden de los nucleótidos de un fragmento de ADN. Es eso de: ATTCGGCCT pero mucho más largo. Para secuenciar necesitamos un poco de material genético o por contra todo el genoma. El Genoma no es otra cosa que la información genética que posee un organismo (o sus mitocondrias). La Leucemia Linfática Crónica (LLC) es una enfermedad de la sangre que afecta a los glóbulos blancos. No está provocada por un virus, una bacteria o un protozoo. No hay un “ente biológico” susceptible de ser capturado, estudiado y secuenciado (como sí que se puede hacer con el SIDA o el Chagas por poner un par de ejemplos). Decir que se ha secuenciado la leucemia es exactamente lo mismo que decir que se ha secuenciado la calvicie.[…]
Además, en el blog de Emilio Cervantes (Científico Titular del CSIC (Consejo Superior de Investigaciones Científicas) en el Instituto de Recursos Naturales y Agrobiología de Salamanca) titulado Biología y pensamiento hace mención a lo mismo que se escribe aquí y en «La muerte de un ácaro». Citaré una parte del artículo más que aclaratoria:
[…]El ICGC es un consorcio internacional dedicado al estudio del cáncer en el que participan grupos de investigación de diversos paises. Según explica en su web, y en un detallado artículo en la revista Nature, su objetivo es lanzar y coordinar un gran número de proyectos de investigación para esclarecer los cambios presentes en los genomas de pacientes que presentan diversas formas de cáncer. (Tales cambios en los genomas pueden ser, como en el artículo indicado más abajo, lo que siempre se llamó mutaciones, y afectan a determinados genes o secuencias particulares del genoma; ni una mutación, ni un cambio constituye un genoma nuevo). Algunos grupos españoles participan en particular en el análisis de la Leucemia linfocítica crónica (CLL) y su objetivo explicado en la web es obtener un catálogo completo de las alteraciones genéticas en 500 tumores independientes. A tal fin se ha publicado recientemente en Nature el artículo titulado Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia.
Es un disparate indicar como la mayoría de las noticias vienen haciendo que se haya secuenciado el genoma de la Leucemia. La leucemia es una enfermedad y como tal carece de genoma.
Lo que se ha hecho es secuenciar el genoma de pacientes con leucemia identificándose ciertas mutaciones. Algunas de ellas se asocian con determinados genes.[…]
[…]Pero convendría también recordar que ningún genoma ni humano, ni de metazoos ha sido secuenciado en su totalidad. En todos los proyectos de secuenciación de genomas existen huecos correspondientes a los centrómeros, heterocromatina y otras regiones ricas en secuencias repetidas.
El trabajo publicado y al que hacen referencia las noticias consiste en el primer análisis exhaustivo de la CLL, combinando la secuenciación del genoma con características y resultados clínicos. Se destaca la utilidad de este enfoque para la identificación de mutaciones clínicamente relevantes en el cáncer.
No se secuencia el genoma de la leucemia. La leucemia no tiene genoma.[…]
Después de exponer todo lo anterior, creo que queda claro tanto la definición de GENOMA como el por qué de el error en la afirmación de los titulares.
He podido saber que todo fue un cúmulo de situaciones y el error ya está subsanado, como se puede comprobar en la nueva nota de prensa del CSIC y CNB.
Espero que, como me ha pasado a mi, lo que realmente haya llegado a la gente no sea este error sino el avance científico que supone la detección de nuevas mutaciones en genes responsables de un cáncer. Eso es lo que importa.
[Nota: Quiero agradecer al equipo de Noticias del CSIC y CNB por ayudar a esclarecer los hechos y demostrar su gran profesionalidad para asegurarse de que se haya divulgado correctamente el estudio sobre la leucemia linfocítica crónica]
Esta afirmación, publicada en numerosos artículos a lo largo y ancho del planeta, es demasiado tajante. Luego os explico por qué. Pero no deja de ser cierta al día de hoy. Se trata del animal con el mayor número de genes contabilizado gracias a la secuenciación de su genoma. Se trata de un crustáceo pequeño cuyo nombre científico es Daphnia pulex y que se le conoce vulgarmente como pulga de agua. Además de ser el animal con una mayor cantidad de genes -31.000 frente a los cerca de 23.000 que tiene el ser humano- también es el primer crustáceo que ha sido secuenciado su genoma en su totalidad.
La causa de que este pequeño animal tenga esa cantidad desmesurada de genes se debe a que éstos están copiados varias veces a lo largo de su genoma. Esa duplicación es tres veces más alta de lo normal y supone un 30% más que lo que sucede en nuestro genoma.
Existe una web asociada a la investigación genómica específica de esta Pulga de agua: el Daphnia Genomics Consortium. Aquí hay una muy buena cantidad de información que puede ser de ayuda para otros estudios genómicos. Desde los proyectos desarrollados hasta las publicaciones y protocolos básicos a seguir en los estudios genómicos. Incluso se enlazan las bases de datos y fuentes asociadas a este crustáceo. Es bastante impresionante lo que hay detrás de una cosa tan pequeña.
Volviendo a su genómica, se ha observado que «más de un tercio de los genes de Daphnia no están documentados previamente. Por lo tanto son nuevos para la Ciencia» dice Don Gilbert, co-autor y científico del Departamento de Biología de la Universidad de Indiana.
Pero lo verdaderamente interesante no está en la cantidad. Si no en la cualidad de los genes. Este crustáceo tiene la capacidad de variar su expresión génica dependiendo del estrés que sufre. No es algo único de estos seres. Lo que sucede es que esta pulga puede ser utilizada fácilmente como modelo para poder analizar la expresión de esos genes y extrapolar esa información a otros organismos. Incluido los seres humanos, ya que no somos tan distintos al fin y al cabo.
Un ejemplo de sus habilidades es la capacidad de adaptarse a las contaminaciones en su ambiente acuático. Dependiendo de la presencia o ausencia de determinados compuestos químicos -en cantidades muy pequeñas para su medición- su expresión génica varía. Se ha postulado la posibilidad de utilizar estos animales como marcadores de esa contaminación en el ambiente, pudiéndose reducir los costes mediante el estudio de su expresión en comparación con los métodos utilizados hasta ahora. Por tanto es un modelo que sirve para observar la integridad de los ecosistemas acuáticos.
Pero ese parecido genético con nosotros también permite -según dice Joseph Shaw, también co-autor del estudio, científico del Departamento de Biología de la Universidad de Indiana y especialista en Ecología- relacionar estos conocimientos asociados con la calidad de las aguas con enfermedades del ser humano.
El estudio de los genes duplicados que comenté anteriormente también tiene su gran importancia. Tanto a nivel evolutivo, ya que una duplicación de genes permite una preservación más fácil de la especie, como de la expresión génica adyacente de otros genes, ya que se propone que tantos genes duplicados pueden inferir su acción en otras cascadas de expresión génica de otros genes. No quiero aburrir con el interminable tema de la regulación y expresión génica.
Terminando quería subrayar el tema del título del artículo y que se ha pasado por alto en numerosas publicaciones (en la publicación original son bien visibles las interrogaciones que dejan abierta la afirmación): no se puede dictaminar tan tajantemente sobre la cualidad de que sea el animal con el mayor número de genes. Primero porque estamos en pañales del conocimiento genómico. Sabemos las piezas pero no cómo funcionan TODAS por separado y en conjunto para comprender la maquinaria que compone un ser vivo. Y este es un organismo modelo y que, por tanto, representa a otros muchos. Pero aunque sean parecidos no tienen por qué tener el mismo número de genes. Y visto el tema de la duplicación génica que es el que da el número tan elevado de genes, es de cajón pensar que habrá muchos organismos que tengan más genes. Aunque no sean modelo.
El pasado 8 de Diciembre, en la revista Science Translational Medicine se publicó cierto avance sobre el diagnóstico de enfermedades genéticas gracias a la obtención del genoma del feto mediante al análisis de la sangre de la madre. El estudio revela que hay una cierta cantidad de ADN del futuro hijo en el plasma de la sangre materna. Este ADN está degradado y por lo tanto son multitud de fragmentos «flotando» por ese suero sanguíneo.
Las avanzadas técnicas de secuenciación que ya hemos comentado en el blog, permiten el análisis de una gran cantidad de fragmentos y los programas bioinformáticos permiten ordenar esas secuencias. Los estudios para desenmascarar ese ADN del feto son muy exhaustivos y costosos (por ahora), pero se espera que en un futuro no muy lejano los costes se abaraten y sea una prueba que permita eliminar métodos invasivos como la amniocentesis para detectar enfermedades hereditarias.
El estudio se comprobó mediante un análisis a una pareja que podría dotar a su todavía no nato hijo la enfermedad de la beta-talasemia. Gracias a la comparativa del ADN de la madre y del padre junto con el obtenido del plasma sanguíneo comprobaron que la técnica permitía averiguar con certeza si el hijo tendría esa herencia perjudicial para su salud. Recordando un poco de genética, ese gen causante de la beta-talasemia está envuelto en la formación de la hemoglobina y que provoca un bajo rendimiento en la toma de oxígeno. Su herencia es clásica (si los padres son portadores del alelo mutado o malo) con un 25% de probabilidades de que herede los dos alelos «buenos», un 50% de que herede con ambos alelos sin causar la enfermedad y un 25% de probabilidades de que lleve ambos alelos malos y desarrolle la enfermedad. El estudio del genoma del feto por medio del análisis del plasma sanguíneo permite obtener unas proporciones de los genes de ambos genomas, de la madre y del niño. Estudiando esa proporción se puede estimar si el niño lleva o no la carga genética buena o la mala. Mediante el estudio con la amniocentesis se corroboró todo y se pudo asegurar que el futuro hijo nacerá sano. En el trabajo se pudo secuenciar e identificar el 94% del genoma del feto para su comparación final de más de 900000 puntos a contrastar.
He podido leer en varios periódicos, como la Vanguardia, que explican el estudio pero verdaderamente no lo aclaran, llevando a la confusión a los lectores que comentaban cosas como que se podía conocer todo ya que las células sanguíneas dan toda la información como el cariotipo (esquema de la colocación e identificación de los cromosomas gracias a encontrarse en la fase metafase). Lo que realmente importa es que el ADN del feto que se obtiene está disgregado en el PLASMA. No hay células sanguíneas completas del feto para hacer un estudio cromosómico. Que se pueda deducir una enfermedad del tipo Síndrome de Down (trisomía del cromosoma 21) por una estimación de los genes que están contenidos en ese cromosoma 21 es una cosa y otra es obtener el cariotipo.
Como siempre, las noticias científicas no son informadas con el rigor que se debería. Yo ya he tomado cartas en este asunto ya que me parece un grandísimo avance. Ya me gustaría que todos mis compañeros investigadores cada vez que vieran faltas de rigor lo comunicaran. Así los periodistas tampoco estarían tan mal valorados.
Un varón japonés no identificado se ha unido al grupo de humanos cuyo genoma ya ha sido secuenciado en su totalidad desde 2001. Los otros seis individuos son el genoma de James Watson, que co-descubrió la estructura del ADN, Craig Venter, el magnate de los EE.UU. de la biotecnología, un hombre de la etnia Yoruba de África occidental, dos hombres coreanos, y un varón de etnia china Han.
El estudio, publicado en la revista especializada Nature Genetics, está encabezado por Tatsuhiko Tsunoda del Centro de Medicina Genómica de Yokohama.
Un consorcio internacional de investigación ha puesto en marcha el denominado «Proyecto Mil Genomas» (Thousand Genomes Project), dirigido a la secuenciación completa del genoma de 1.000 personas anónimas y publicar los datos en el dominio público.
El proyecto tiene como objetivo arrojar luz sobre las variaciones genéticas que pueden explicar la vulnerabilidad sobre enfermedades hereditarias y poder adaptar los medicamentos a las necesidades individuales.
Tsunoda dijo que estaba cauteloso acerca de hacer una comparación rápida entre los japoneses y los otros genomas conocidos. «Más muestras -decenas- sería necesario, que es nuestro plan de futuro», dijo.
Tsunoda dijo que su equipo estaá trabajando en nuevas maneras de detectar patrones de múltiples variaciones en el código genético.
«En el futuro, seremos capaces de encontrar gran número de variaciones en los genomas individuales que deberían estar relacionadas con muchas enfermedades», dijo Tsunoda.
Al paso que van los avances en secuenciación, no tengo duda que en un futuro cercano podremos pedir que nos impriman una copia de nuestro ADN como si el informe de vida laboral se tratara. Espero que os haya gustado.
Vaya vaya. Parece ser que la genética vuelve a revolucionar la medicina. Por primera vez en la historia, un estudio de secuenciación de ADN ha hecho cambiar el diagnóstico de los médicos. Se trata de un caso en Turquía en el que un niño de pocos meses padecía un trastorno que provocaba pérdida de peso y deshidratación. Al principio, los médicos diagnosticaron como causa más probable el síndrome de Bartter. Es un mal que afecta a uno de cada 100000 bebés y causa bajos niveles de potasio y otras sales ocasionando un fallo en el riñón. Menos mal que, para completar el estudio, enviaron muestras de sangre al genetista Richard Lifton del Yale Medical School. De esta forma querían ratificar cuál era el gen causante de la enfermedad. Pero Lifton sospechó de que no era ese síndrome el culpable de la enfermedad del niño y lo que hizo fue secuenciar el genoma del bebé. Esto hubiera sido impensable hace años por los costes, el tiempo y la tecnología a utilizar, pero ahora es otro cantar. Pues menuda sorpresa se llevaron todos al comprobar que el síndrome de Bartter no era la causa sino otro trastorno llamado «congenital chloride diarrhea», que provoca también bajos niveles de sales.
El caso ha sido publicado en Proceedings of the National Academies of the Sciences (PNAS, para los que estamos muy metidos) el pasado octubre. Para que os hagáis una idea, el coste de descifrar 6 mil millones de bases en el genoma humano a caído de 1 million de dólares en 2007 a menos de 20000 dólares hoy en día. Lifton utilizó un método de extracción de dos pasos y secuenció sólo el 1% de esas bases, que contienen genes conocidos, disminuyendo el coste a 2500 dólares.
Como podéis deducir, una nueva era médica basada en la genética está comenzando a llegar. Antes no se aprobaba el diagnóstico por secuenciación. Ya no sólo por la tecnología, sino por los acuerdos con los seguros médicos.
Realmente, noticias como esta hacen que me guste cada vez más mi profesión.
Fuente: Genetic diagnosis by whole exome capture and massively parallel DNA sequencing
Este sitio web utiliza cookies para que usted tenga la mejor experiencia de usuario. Si continúa navegando está dando su consentimiento para la aceptación de las mencionadas cookies y la aceptación de nuestra política de cookies, pinche el enlace para mayor información.
ACEPTAR