Las enzimas de restricción
A lo largo de estas semanas estoy volviendo a trabajar con enzimas de restricción y, antes de publicar un artículo sobre ciertas aplicaciones, será buena idea explicar un poco sobre el tema.
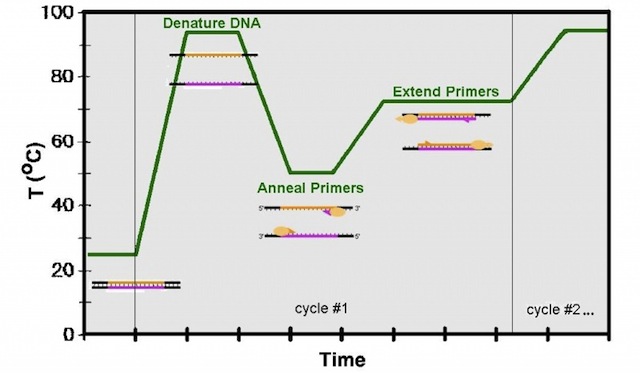
Las enzimas (ó endonucleasas) de restricción son unas proteínas con acción catalítica que tienen la propiedad de reconocer secuencias cortas, de unos cuatro ó seis pares de bases en el ADN de doble hebra, y cortar en todos los puntos donde se encuentre dicha secuencia. Este lugar de corte se denomina diana de restricción. Cada enzima de restricción tiene una diana particular en el ADN. En la actualidad hay un amplísimo catálogo de enzimas de restricción (podéis ver un ejemplo en el listado de Takara) y los usos destinados son varios: para la realización de mapas físicos (hablamos de ADN, no de cordilleras XDD), huella genética ó fingerprinting, búsqueda de polimorfismos tipo SNPs, AFLPs, RFPLs, utilización en pasos previos a la clonación…etc. Y multitud de aplicaciones más específicas que hacen muy versátiles en su uso.
Con un ejemplo queda todo más claro: una enzima muy típica es Alu I que reconoce la secuencia AGTC y corta la doble hélice del ADN entre la Guanina y la Timina. Su actuación sería darse un paseo por toda la hebra de ADN en búsqueda de esa diana. En el momento que la encuentra, se ancla y corta, obteniéndose dos fragmentos. Si no hubiera esa secuencia diana, no corta y el fragmento de ADN de doble cadena que teníamos quedará igual que antes. Si os dais cuenta, es un buen método para diferenciar individuos. La variación de nucleótidos entre distintos individuos puede observarse de esta forma sin necesidad de secuenciar.
No se aplican estas enzimas a la totalidad del ADN genómico, ya que se obtendrían multitud de bandas que no se podrían distinguir bien. Se amplifican, mediante PCR, ciertos lugares específicos del ADN para que las dianas de restricción permitan obtener unos resultados claros para su posterior análisis.
Para los que trabajamos en un laboratorio esto que acabo de explicar es como usar el cuchillo en las comidas, pero ya he visto que hay cierto interés en ciertas aplicaciones de uso en el laboratorio y pronto haré una review de herramientas que facilitan el estudio mediante enzimas de restricción.
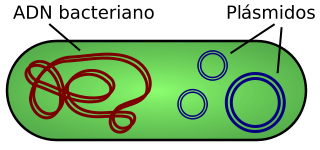
Por ahora, para que no sea un rollo lo que he explicado, os dejo con un vídeo (es una animación en 3D) sobre como actúa una enzima de restricción para clonar un fragmento de ADN. Recordad lo que es la clonación. Es una de las técnicas de ingeniería genética más comunes en los laboratorios de biología molecular. Se puede ver como la actuación de la enzima proporciona unos extremos en el plásmido (son moléculas de ADN extracromosómico circular o lineal que se replican y transcriben independientes del ADN cromosómico.  Están presentes normalmente en bacterias, y en algunas ocasiones en organismos eucariotas como las levaduras. Su tamaño varía desde 1 a 250 kb. El número de plásmidos puede variar, dependiendo de su tipo, desde una sola copia hasta algunos cientos por célula) que son complementarios al corte hecho a un fragmento de ADN que queremos insertar con la misma enzima de restricción. Por último serán unidos los extremos por otra enzima llamada ligasa. Así se puede incluir como si formara parte del plásmido y obtener copias idénticas del fragmento de interés al replicarse la bacteria.
Están presentes normalmente en bacterias, y en algunas ocasiones en organismos eucariotas como las levaduras. Su tamaño varía desde 1 a 250 kb. El número de plásmidos puede variar, dependiendo de su tipo, desde una sola copia hasta algunos cientos por célula) que son complementarios al corte hecho a un fragmento de ADN que queremos insertar con la misma enzima de restricción. Por último serán unidos los extremos por otra enzima llamada ligasa. Así se puede incluir como si formara parte del plásmido y obtener copias idénticas del fragmento de interés al replicarse la bacteria.
[youtube]http://www.youtube.com/watch?v=yDGA8n1oJ5Q[/youtube]
Espero que os haya gustado.