La nueva colaboración en Trending Ciencia, el podcast científico de moda, trata del último gran bombazo presentado en la revista Nature y publicado en su versión online el pasado 7 de mayo donde se explica la generación de un organismo semi-sintético en el que se ha ampliado su alfabeto genético añadiendo un par de bases nitrogenadas distintas a las cuatro del ADN (adenina, Guanina, Citosina y Timina).
Para terminar el programa de 7 minutos, una mención a la innovadora idea de utilizar el ADN como soporte digital. ¿Os imagináis cambiar los almacenamientos de información actuales por moléculas de ADN?.
Un programa que seguro no dejará a nadie indiferente.
No dudéis en comentar este episodio en esta misma entrada en la web o mediante las redes sociales mencionando nuestra cuenta oficial de Twitter @TrendingCiencia o personalmente al autor del programa, que soy yo,@DoctorGenoma. Y animad a todo al que le guste la ciencia a suscribirse a nuestro podcast mediante su FEED o a través de las plataformas iTunes e iVoox.
El mes pasado se pudo conocer los resultados de un estudio realizado en tierras camboyanas donde se muestran datos esclarecedores de cómo los periodos de inanición provocados por las situaciones bélicas pueden desencadenar que nuestra genética sea peligrosamente modificada.
Actualmente la diabetes está siendo un gran problema en Camboya. En realidad no es algo sorpresivo. Ahora mismo se come mucho más, la gente tiene una vida media mayor y se realiza mucho menos ejercicio físico que en épocas anteriores. Sin embargo ciertos investigadores han estudiado este incremento y piensan que hay algo más detrás de un cambio en los hábitos y de la esperanza de vida.
Kim Keuky atendiendo a varias mujeres camboyanas diabéticas. Foto: Kounila Keo.
Desde 1975 a 1979 Camboya fue gobernada por los Jemeres Rojos, régimen totalitario que acabó con la vida de entre 700000 y 2000000 de personas debido a los asesinatos y los muertos ocasionados por el hambre. El endocrinólogo Lim Keuky piensa que esos periodos de inanición han tenido gran culpa de los diagnósticos de diabetes que está observando en pacientes rondando los 30 años, edad muy temprana para desarrollar una diabetes de tipo 2 y más aún en el lugar donde le ha llamado la atención: una aldea rural donde viven muchos descendientes de las personas que sobrevivieron a las salvajadas de los Jemeres Rojos. Incluso, cuando viaja a otros países desarrollados y cuenta sus observaciones no le creen: «Cuando voy al extranjero a los países desarrollados, la gente dice que estoy mintiendo. No estoy mintiendo «, declara el endocrinólogo.
Pero es que esto no es un caso aislado de un país de oriente. Otro ejemplo lo tenemos más cerca, en occidente. Los investigadores en los Países Bajos, han notado un fenómeno similar. Ellos han estado estudiando a los supervivientes del «invierno del hambre», un oscuro episodio durante la ocupación nazi de la Segunda Guerra Mundial.
En el invierno de 1944 a 1945, los nazis habían bloqueado todos los alimentos que llegaran a la zona occidental de los Países Bajos. La Doctora Rebecca Painter del Centro Médico Académico de Amsterdam dice que «la gente estaba recibiendo menos de 1.000 calorías al día».
Painter y sus colegas decidieron hacer un seguimiento de una hipótesis de un médico inglés, David Barker, que tenía la teoría de que las condiciones en el útero pueden tener efectos permanentes en el desarrollo fetal.
Los investigadores holandeses examinaron muestras de sangre de las personas nacidas en la región afectada por el invierno del hambre. Encontraron que los nacidos durante la hambruna fueron significativamente más propensos a desarrollar diabetes o pre-diabetes a los 50 años en comparación con los nacidos sólo un año antes o después. La hipótesis de Barker se denominó como el «fenotipo ahorrativo». Los fetos en desarrollo en las madres que están recibiendo apenas lo suficiente para comer tienen que adaptarse. «Se adaptan de una manera que los pone en un mayor riesgo de desarrollar diabetes en la edad adulta «, explicaba David Baker . Con una toma insuficiente de nutrientes que viajan a través de la placenta, el páncreas del feto (que produce la insulina que previene la diabetes) no se desarrolla correctamente, dice la Doctora Painter.
Painter cree que el proceso está en el trabajo en Camboya. «Hay una buena probabilidad de que la inanición que Camboya vio a finales de 1970 puede contribuir a su actual aumento de la diabetes», dice la Doctora.
Lim Keuky sospecha que el aumento de la diabetes se ha visto agravada por el hecho de que la disponibilidad de alimentos en Camboya es mucho mejor hoy que en años anteriores. Afirma que «Ahora tenemos mucho que comer y el páncreas no puede soportar la comida todos los días». Además, dice que Camboya debe prepararse para un mayor número de casos de diabetes en los próximos años. Muchos camboyanos ignoran los primeros síntomas de la diabetes, tales como la sed o hambre excesiva, fatiga y adormecimiento en las manos o los pies. A la larga, la falta de tratamiento conduce a daños en los órganos que pueden ocasionar derrames cerebrales, insuficiencias cardíacas o insuficiencias renales, que pueden ser fatales.
Los expertos comentan ante tales evidencias que otros países en desarrollo, que han experimentado períodos de hambre, deben estar en la búsqueda de picos similares en diabetes. Investigadores en China informaron recientemente que los niños nacidos allí durante el período de tiempo de hambruna entre los años 1959 y 1961 eran más propensos a desarrollar diabetes en el futuro que las personas nacidas antes o después. También se han recibido informes similares de África meridional.
Pero esto llega a nivel genético. Los estudios en animales plantean una posibilidad aún más preocupante: «Hay un efecto multigeneracional», dice la Doctora Painter . En otras palabras, los cambios en el funcionamiento de nuestros genes, que son causados por la hambruna, pueden transmitirse a las generaciones futuras. ¿Recordáis la epigenética? Recientemente se ha demostrado que algunos cambios epigenéticos son heredables. Eso significa que los camboyanos nacidos mucho después de aquel régimen de los Jemeres Rojos también pueden sufrir efectos físicos de aquellos años de brutalidad.
En esta aparición en el programa de divulgación científica Trending Ciencia, podéis escucharme comentando la noticia de hace unos días referente a la utilización de las tecnologías de clonación por parte del gobierno Chino dirigidas al ganado porcino en pos de la investigación genética. Además, os pondréis al tanto del potencial de este gigante asiático en el campo de la secuenciación y su objetivo de clonar lo que llaman «animales adorables».
No dudéis en comentar este episodio en esta misma entrada en la web o mediante las redes sociales mencionando nuestra cuenta oficial de Twitter @TrendingCiencia o personalmente al autor del programa, que soy yo,@DoctorGenoma. O es de más animar a todo el que le guste la ciencia a suscribirse a nuestro podcast mediante su FEED o a través de las plataformas iTunes e iVoox.
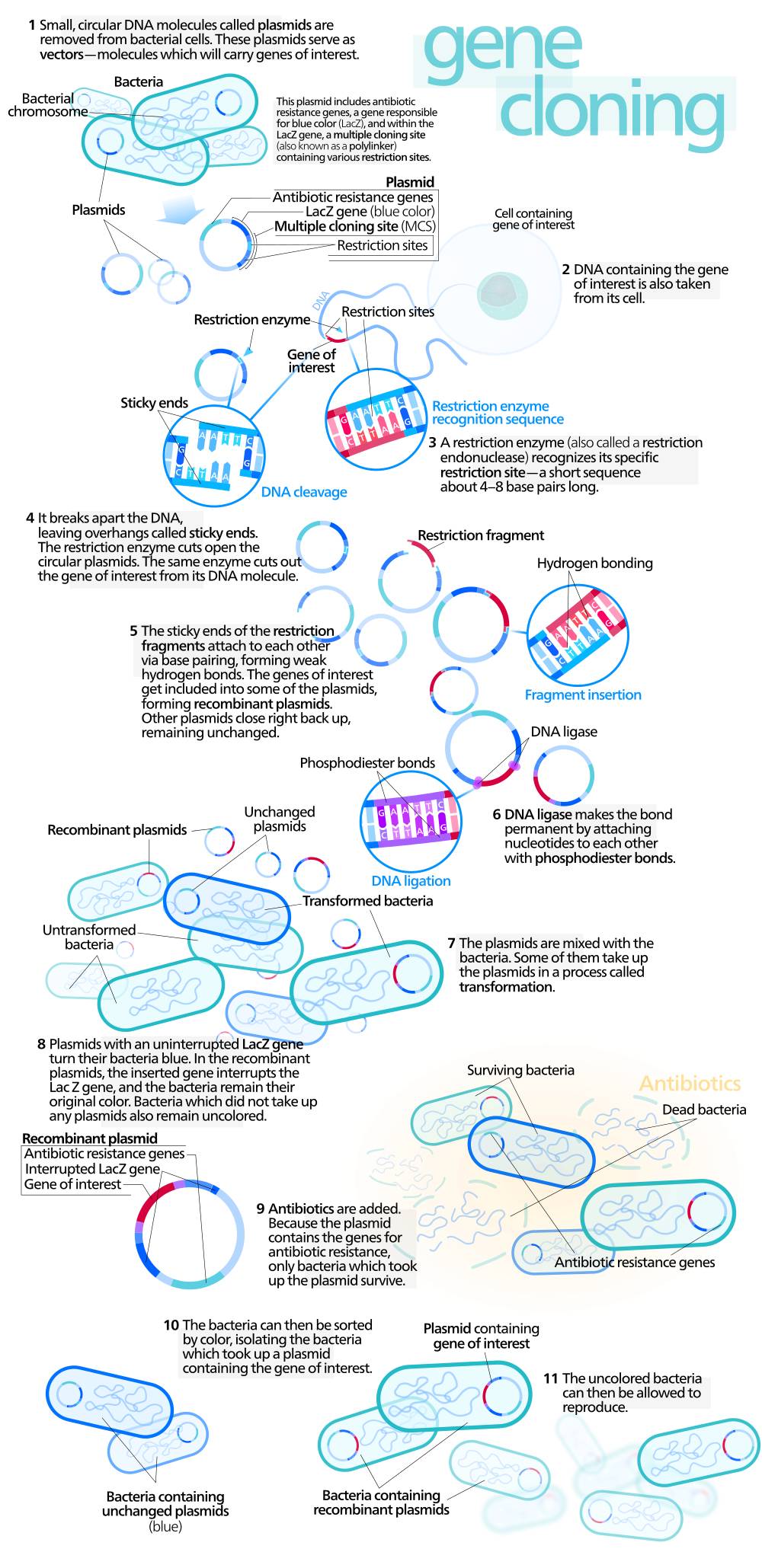
Se acaba de publicar un nuevo método de ingeniería genética que reduce drásticamente el tiempo y el esfuerzo necesarios para insertar genes exógenos en las bacterias, los caballos de batalla de la biotecnología. El artículo se encuentra en la revista ACS Synthetic Biology, y este novedoso estudio abre el camino para un desarrollo más rápido de los microorganismos «de diseño» que pueden utilizarse para el desarrollo de medicamentos, limpieza ambiental y muchas otras actividades.
Keith Shearwin y sus colaboradores explican que la colocación, o la integración, de una porción de ADN en el genoma de una bacteria es crítico para la obtención de bacterias de diseño. Ese ADN puede dar la capacidad a los microbios de resistencia ante ciertos antibióticos, por ejemplo, o la capacidad de generar sustancias que degraden el crudo después de un gran vertido. Esquema de un modelo de Clonación de ADN. Pulsar para ampliar.Sin embargo, los métodos actuales de ingeniería genética son costosos tanto en tiempo como en multitud de pasos a realizar para llevarlos a cabo. Para hacer frente a estos inconvenientes, los investigadores han desarrollado una nueva tecnología de la ingeniería genética de un solo paso, al que llamaron «clonetegration» en referencia a los clones o copias de genes o fragmentos de ADN.
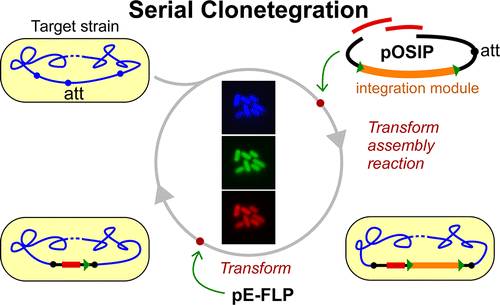
Los científicos, perteniecientes a un grupo de investigación combinado por la Universidad de Stanford (California, Estados Unidos) y la Universidad de Adelaida (Australia), describen el desarrollo de la técnica y su éxito en las pruebas de laboratorio realizadas mediante el método de «clonetegration» en bacterias E. coli y Salmonella typhimurium, muy utilizadas en biotecnología.
«El método es rápido, eficiente y fácil de hacer y se pueden integrar varios genes al mismo tiempo«, Dicen los investigadores. Además predicen que clonetegration «se convertirá en una técnica valiosa facilitar la ingeniería genética con secuencias difíciles de clonar y rápida construcción de los sistemas biológicos sintéticos.»
Esquema de la «Clonetegration» seriada.
Espero que pronto pueda tener acceso a este artículo y poder estudiar más detenidamente el proceso que proponen, porque mi curiosidad sobre este nuevo sistema de clonación no me va dejar dormir. Por ahora, empezaré a leer este suplemento de la publicación para ver si me convence.
Referencia «One-Step Cloning and Chromosomal Integration of DNA,» ACS Synthetic Biology. DOI: 10.1021/sb400021j
Hace unos días recibí una propuesta de Adrián, el responsable del blog Científicamente Correcto : hacerme una entrevista. ¡ Menuda sorpresa!. Sobre todo tras mi reciente doctorado. Estos detalles suben la moral científica ( y la no científica también ;D). Y más por el momento difícil por el que está pasando la investigación española.
En fin, si queréis conocerme un poco más os animo a que leáis esta entrevista. Os pongo el enlace a continuación. Y ya me contaréis que os ha parecido 😉
.
Después del largo camino, aquí me tenéis. Cambiando en todos los currículos, webs, redes y perfiles aquello de «estudiante de doctorado». Ya soy Doctor. Bueno, como bien se dice, no se es Doctor hasta que no se pagan las tasas. Pero todo lo que tenía que hacer científicamente, ya está hecho. Si me seguís por las redes sociales, habréis podido leer aquello de «Paseando con traje y corbata sabiendo que soy DOCTOR con papeles. Indescriptible». Todos los que han conseguido el grado, lo han sentido. En mi caso, mezcla de orgullo por la valoración de los expertos, satisfecho por como salió todo, contento por haber demostrado ante amigos y familia lo que se ha realizado durante todo este tiempo, agradecido por ciertos detalles de compañeros de departamento…todo ello mezclado con cierta inquietud por terminar una etapa y comenzar otra, muy dura en estos momentos por la mala situación.
Pero sienta bien cumplir el sueño de ser Doctor. Y, aunque algunos vayan diciendo cosas como «ya tenía ganas de terminar»…» se hacía pesado»…»menudo peso me he quitado de encima»…son palabrerías tópicas típicas de cara a la galería. Si son buenos investigadores, no será lo que hayan pensado de verdad. Y si no, debo ser un bicho raro. Un doctorado, se DISFRUTA. Luego, ya vendrán otras cosas 😉
Tras varios años participando y asistiendo a los congresos de la SEG, este año tan sólo puedo animar a su asistencia. Ya van 38 ediciones de este evento que se celebra cada dos años y que, en esta ocasión, la ciudad anfitriona es Murcia. Se celebrará esta semana, entre los días 21 y 23 de septiembre. Os dejo el programa para que le echéis un vistazo.
Desde mi punto de vista, la asistencia a congresos es algo muy rico tanto para pasar un buen rato con conocidos y nuevos amigos como a nivel profesional. Muchos sólo asisten a estos congresos «generales» cuando les interesa. Es algo asumible y de respetar. Pero, de vez en cuando, es bueno ver qué se cuece en otros campos de la Genética.
Por último, desear toda la suerte del mundo a algunos amigos y conocidos que sé que participarán como ponentes en esta edición y que me duele no poder asistir.Bueno, dejo caer la idea de hacer un life-streaming para que podamos disfrutar del congreso, al menos, los socios de la SEG. Ya me contaréis como ha ido.
Web del XXXVIII Congreso de la Sociedad Española de Genética
Esta afirmación, publicada en numerosos artículos a lo largo y ancho del planeta, es demasiado tajante. Luego os explico por qué. Pero no deja de ser cierta al día de hoy. Se trata del animal con el mayor número de genes contabilizado gracias a la secuenciación de su genoma. Se trata de un crustáceo pequeño cuyo nombre científico es Daphnia pulex y que se le conoce vulgarmente como pulga de agua. Además de ser el animal con una mayor cantidad de genes -31.000 frente a los cerca de 23.000 que tiene el ser humano- también es el primer crustáceo que ha sido secuenciado su genoma en su totalidad.
La causa de que este pequeño animal tenga esa cantidad desmesurada de genes se debe a que éstos están copiados varias veces a lo largo de su genoma. Esa duplicación es tres veces más alta de lo normal y supone un 30% más que lo que sucede en nuestro genoma.
Existe una web asociada a la investigación genómica específica de esta Pulga de agua: el Daphnia Genomics Consortium. Aquí hay una muy buena cantidad de información que puede ser de ayuda para otros estudios genómicos. Desde los proyectos desarrollados hasta las publicaciones y protocolos básicos a seguir en los estudios genómicos. Incluso se enlazan las bases de datos y fuentes asociadas a este crustáceo. Es bastante impresionante lo que hay detrás de una cosa tan pequeña.
Volviendo a su genómica, se ha observado que «más de un tercio de los genes de Daphnia no están documentados previamente. Por lo tanto son nuevos para la Ciencia» dice Don Gilbert, co-autor y científico del Departamento de Biología de la Universidad de Indiana.
Pero lo verdaderamente interesante no está en la cantidad. Si no en la cualidad de los genes. Este crustáceo tiene la capacidad de variar su expresión génica dependiendo del estrés que sufre. No es algo único de estos seres. Lo que sucede es que esta pulga puede ser utilizada fácilmente como modelo para poder analizar la expresión de esos genes y extrapolar esa información a otros organismos. Incluido los seres humanos, ya que no somos tan distintos al fin y al cabo.
Un ejemplo de sus habilidades es la capacidad de adaptarse a las contaminaciones en su ambiente acuático. Dependiendo de la presencia o ausencia de determinados compuestos químicos -en cantidades muy pequeñas para su medición- su expresión génica varía. Se ha postulado la posibilidad de utilizar estos animales como marcadores de esa contaminación en el ambiente, pudiéndose reducir los costes mediante el estudio de su expresión en comparación con los métodos utilizados hasta ahora. Por tanto es un modelo que sirve para observar la integridad de los ecosistemas acuáticos.
Pero ese parecido genético con nosotros también permite -según dice Joseph Shaw, también co-autor del estudio, científico del Departamento de Biología de la Universidad de Indiana y especialista en Ecología- relacionar estos conocimientos asociados con la calidad de las aguas con enfermedades del ser humano.
El estudio de los genes duplicados que comenté anteriormente también tiene su gran importancia. Tanto a nivel evolutivo, ya que una duplicación de genes permite una preservación más fácil de la especie, como de la expresión génica adyacente de otros genes, ya que se propone que tantos genes duplicados pueden inferir su acción en otras cascadas de expresión génica de otros genes. No quiero aburrir con el interminable tema de la regulación y expresión génica.
Terminando quería subrayar el tema del título del artículo y que se ha pasado por alto en numerosas publicaciones (en la publicación original son bien visibles las interrogaciones que dejan abierta la afirmación): no se puede dictaminar tan tajantemente sobre la cualidad de que sea el animal con el mayor número de genes. Primero porque estamos en pañales del conocimiento genómico. Sabemos las piezas pero no cómo funcionan TODAS por separado y en conjunto para comprender la maquinaria que compone un ser vivo. Y este es un organismo modelo y que, por tanto, representa a otros muchos. Pero aunque sean parecidos no tienen por qué tener el mismo número de genes. Y visto el tema de la duplicación génica que es el que da el número tan elevado de genes, es de cajón pensar que habrá muchos organismos que tengan más genes. Aunque no sean modelo.
La uva es uno de los cultivos frutales más demandados en estos momentos. Los estudios de mejora para perfeccionar las uvas son cruciales a la hora de ofrecer al agricultor una variedad que sea lo más resistente a enfermedades y estreses bióticos que existan.
Desde que se introdujo el cultivo de la uva en Oriente próximo hace unos 8000 años se han obtenido relativamente pocos cruzamientos entre variedades que permitan una mayor diversidad genética que ocasione una uva «más fuerte» ante problemas que perjudiquen su desarrollo. La vid es una planta perenne que necesita de unos 3 años para que esté madura para dar esas uvas que tanto nos gustan.
Como gran ayuda en la mejora de plantas, Vitis vitifera (nombre científico de la especie de la uva «domesticada») también ha sido utilizada para multitud de estudios mediante marcadores genéticos que permiten conocer más sobre los rasgos de interés de las variedades utilizadas en los cultivares dedicados al consumo humano. Esos marcadores genéticos estarán asociados a caracteres que tengan que ver con la resistencia o susceptibilidad hacia estreses tanto bióticos (imaginad una plaga de insectos) como abióticos (un ejemplo de ellos serían las sequías). Al conocer esos marcadores y la distribución entre las variedades, permite mejorar la producción de la uva.
En un estudio realizado en Estados Unidos y publicado este pasado 18 de Enero en PNAS, se estudiaron unas 950 muestras de uva mediante un chip de ADN que permite examinar unos 9000 patrones de variabilidad mediante el estudio de SNPs (Polimorfismos de un único nucleótido y de los que hemos hablado en varios artículos anteriormente). Gracias a esos microarrays, los científicos desarrollaron una tabla que describe el parentesco genético de docenas de las accesiones de uva que producen algunos de los vinos más populares del mundo, incluidas uvas como Reisling o Pinot noir que son de gran interés para la producción de vino.
Los resultados del estudio fueron muy curiosos. Desde siglos pasados cuando una uva presentaba un rasgo que las distinguía de otra supuesta variedad de uva y que era beneficioso para el agricultor, se propagaba de forma vegetativa y era asignado un nuevo nombre por ese agricultor. Hay unas 10000 denominaciones de este tipo en todo el mundo actualmente, pero varias de las variedades nombradas por esa mutación encontrada y nombrada como nueva por ello son identicamente genéticas a sus parentales. O, al menos, las pruebas genéticas no pueden distinguir entre los descendientes mutantes y los parentales usados para el cruzamiento.
Sean Myles, estudiante post doctoral en la Escuela de Medicina de la Universidad de Stanford, y sus colegas advierten que el 58% de las 950 accesiones de Vitis examinadas están tan relacionadas que parecen ser clones de una única accesión. De esta forma parece que no hay tanta variabilidad como parece, al menos desde el punto de vista genético. Habría que comparar el estudio con lo que observan los sumilleres al catar el vino.
Referencias: S Myles, A. R Boyko, C. L Owens, P. J Brown, F Grassi, M. K Aradhya, B Prins, A Reynolds, J.-M Chia, D Ware, C. D Bustamante, E. S Buckler.(2011) Genetic structure and domestication history of the grape. Proceedings of the National Academy of Sciences pp. 6
El pasado 8 de Diciembre, en la revista Science Translational Medicine se publicó cierto avance sobre el diagnóstico de enfermedades genéticas gracias a la obtención del genoma del feto mediante al análisis de la sangre de la madre. El estudio revela que hay una cierta cantidad de ADN del futuro hijo en el plasma de la sangre materna. Este ADN está degradado y por lo tanto son multitud de fragmentos «flotando» por ese suero sanguíneo.
Las avanzadas técnicas de secuenciación que ya hemos comentado en el blog, permiten el análisis de una gran cantidad de fragmentos y los programas bioinformáticos permiten ordenar esas secuencias. Los estudios para desenmascarar ese ADN del feto son muy exhaustivos y costosos (por ahora), pero se espera que en un futuro no muy lejano los costes se abaraten y sea una prueba que permita eliminar métodos invasivos como la amniocentesis para detectar enfermedades hereditarias.
El estudio se comprobó mediante un análisis a una pareja que podría dotar a su todavía no nato hijo la enfermedad de la beta-talasemia. Gracias a la comparativa del ADN de la madre y del padre junto con el obtenido del plasma sanguíneo comprobaron que la técnica permitía averiguar con certeza si el hijo tendría esa herencia perjudicial para su salud. Recordando un poco de genética, ese gen causante de la beta-talasemia está envuelto en la formación de la hemoglobina y que provoca un bajo rendimiento en la toma de oxígeno. Su herencia es clásica (si los padres son portadores del alelo mutado o malo) con un 25% de probabilidades de que herede los dos alelos «buenos», un 50% de que herede con ambos alelos sin causar la enfermedad y un 25% de probabilidades de que lleve ambos alelos malos y desarrolle la enfermedad. El estudio del genoma del feto por medio del análisis del plasma sanguíneo permite obtener unas proporciones de los genes de ambos genomas, de la madre y del niño. Estudiando esa proporción se puede estimar si el niño lleva o no la carga genética buena o la mala. Mediante el estudio con la amniocentesis se corroboró todo y se pudo asegurar que el futuro hijo nacerá sano. En el trabajo se pudo secuenciar e identificar el 94% del genoma del feto para su comparación final de más de 900000 puntos a contrastar.
He podido leer en varios periódicos, como la Vanguardia, que explican el estudio pero verdaderamente no lo aclaran, llevando a la confusión a los lectores que comentaban cosas como que se podía conocer todo ya que las células sanguíneas dan toda la información como el cariotipo (esquema de la colocación e identificación de los cromosomas gracias a encontrarse en la fase metafase). Lo que realmente importa es que el ADN del feto que se obtiene está disgregado en el PLASMA. No hay células sanguíneas completas del feto para hacer un estudio cromosómico. Que se pueda deducir una enfermedad del tipo Síndrome de Down (trisomía del cromosoma 21) por una estimación de los genes que están contenidos en ese cromosoma 21 es una cosa y otra es obtener el cariotipo.
Como siempre, las noticias científicas no son informadas con el rigor que se debería. Yo ya he tomado cartas en este asunto ya que me parece un grandísimo avance. Ya me gustaría que todos mis compañeros investigadores cada vez que vieran faltas de rigor lo comunicaran. Así los periodistas tampoco estarían tan mal valorados.
Este sitio web utiliza cookies para que usted tenga la mejor experiencia de usuario. Si continúa navegando está dando su consentimiento para la aceptación de las mencionadas cookies y la aceptación de nuestra política de cookies, pinche el enlace para mayor información.
ACEPTAR